Living with respiratory conditions can be challenging, and understanding the distinctions between different lung diseases is crucial for proper management and treatment. Two conditions that are sometimes confused due to similar names are cystic fibrosis and pulmonary fibrosis. While both affect the lungs, they are distinct diseases with different causes, onset patterns, and treatment approaches.
In this comprehensive guide, we'll explore the key differences between these two conditions, helping you better understand their unique characteristics, symptoms, and management strategies.
Understanding the Basics of Each Condition
What is Cystic Fibrosis?
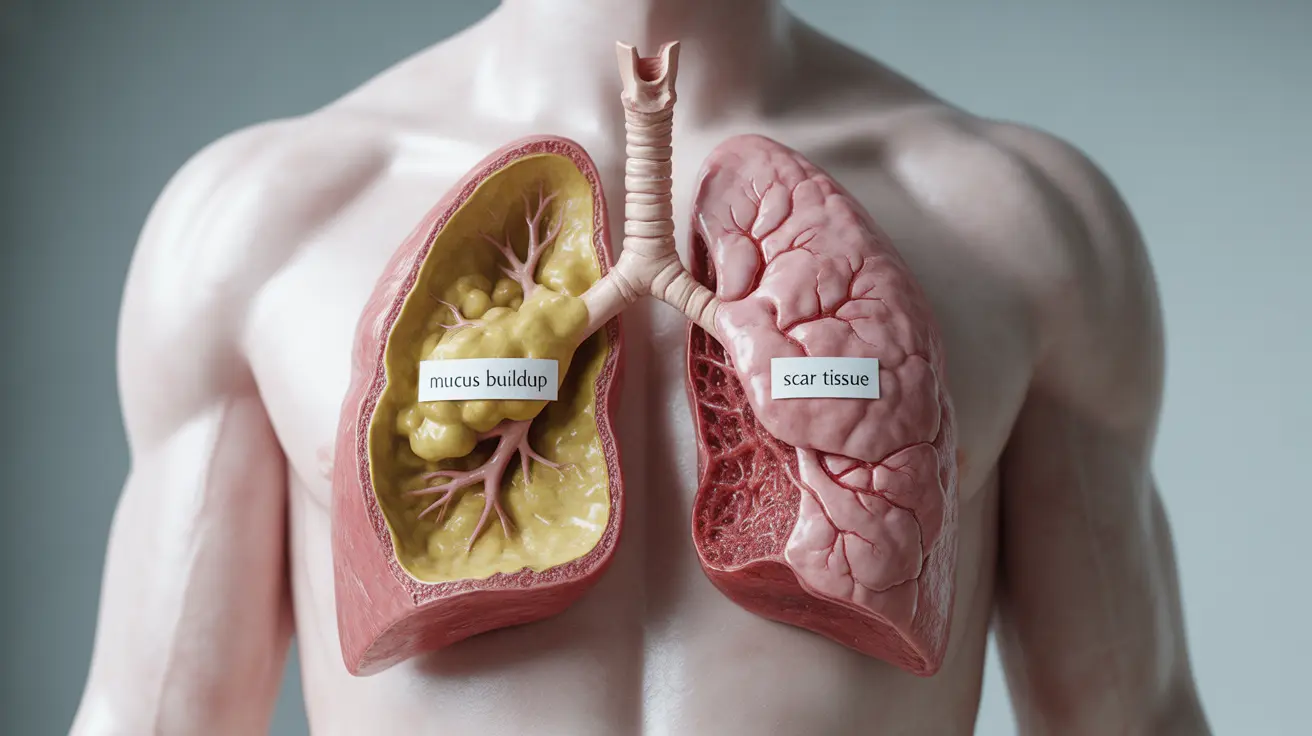
Cystic fibrosis (CF) is a genetic disorder that affects multiple organ systems, primarily the lungs and digestive system. It occurs due to mutations in the CFTR gene, which controls the movement of salt and water in and out of cells. This inherited condition causes thick, sticky mucus to build up in various organs.
What is Pulmonary Fibrosis?
Pulmonary fibrosis (PF) is a lung disease characterized by scarring (fibrosis) of lung tissue. Unlike CF, it's not genetic but typically develops due to various environmental factors, medical conditions, or sometimes without a clear cause (idiopathic pulmonary fibrosis).
Disease Origins and Risk Factors
Cystic Fibrosis Causes
CF is inherited in an autosomal recessive pattern, meaning both parents must carry the defective gene for a child to develop the condition. The disease is present from birth, though symptoms may not appear immediately.
Pulmonary Fibrosis Causes
PF can develop from various factors, including:
- Exposure to environmental toxins
- Certain medications
- Radiation therapy
- Autoimmune diseases
- Chronic inflammatory conditions
Symptom Presentation and Progression
Cystic Fibrosis Symptoms
CF symptoms typically appear in early childhood and include:
- Persistent coughing with thick mucus
- Frequent lung infections
- Poor growth and weight gain
- Salty-tasting skin
- Digestive problems
Pulmonary Fibrosis Symptoms
PF symptoms usually develop gradually in middle-aged and older adults:
- Progressive shortness of breath
- Dry, hacking cough
- Fatigue and weakness
- Unexplained weight loss
- Clubbing of fingertips
Diagnosis and Treatment Approaches
While both conditions affect the lungs, their diagnostic methods and treatment strategies differ significantly. CF requires genetic testing and sweat chloride tests, while PF diagnosis often involves imaging studies and sometimes lung biopsies.
Treatment for CF focuses on managing symptoms through:
- Airway clearance techniques
- Medications to thin mucus
- CFTR modulators
- Enzyme replacement therapy
- Nutritional support
PF treatment typically includes:
- Anti-fibrotic medications
- Anti-inflammatory drugs
- Oxygen therapy
- Pulmonary rehabilitation
- In severe cases, lung transplantation
Frequently Asked Questions
What is the difference between cystic fibrosis and pulmonary fibrosis in terms of causes and symptoms?
Cystic fibrosis is a genetic condition present from birth that affects multiple organs, causing thick mucus buildup. Pulmonary fibrosis is an acquired condition causing lung scarring, typically developing later in life due to environmental factors or underlying conditions.
How are cystic fibrosis and pulmonary fibrosis diagnosed and treated differently?
Cystic fibrosis is diagnosed through genetic testing and sweat chloride tests, with treatment focusing on mucus clearance and CFTR modulators. Pulmonary fibrosis requires imaging studies and is treated with anti-fibrotic medications and oxygen therapy.
Can cystic fibrosis lead to pulmonary fibrosis or lung scarring?
While cystic fibrosis can cause lung damage and scarring over time, this is different from pulmonary fibrosis. The scarring in CF is secondary to chronic inflammation and infection, while PF involves direct scarring of lung tissue.
What are the common complications associated with cystic fibrosis versus pulmonary fibrosis?
CF complications include recurrent lung infections, pancreatic insufficiency, and male infertility. PF complications primarily involve progressive breathing difficulty, right heart failure, and increased risk of lung infections.
At what age do symptoms of cystic fibrosis and pulmonary fibrosis typically appear?
Cystic fibrosis symptoms usually appear in early childhood, often within the first year of life. Pulmonary fibrosis typically develops in middle-aged and older adults, with most cases diagnosed between ages 50 and 70.